Hohe mechanische Belastungen von Bauteilen stellen zunehmende Anforderungen an die zu verwendenden Materialien. Die Auswahl und die belastungsgerechte Anwendung eines Materials werden durch dessen Eigenschaften bestimmt. In der Materialentwicklung wird daher permanent versucht, die Materialeigenschaften an die mögliche Anwendung anzupassen. Das Verständnis der komplexen Verhaltensweisen in der Mikroebene zwischen Atomen und Molekülen ist bedeutend für die Weiterentwicklung des Eigenschaftspotenzials, beispielsweise einer höheren Festigkeit. Eine leistungsfähige Methode für die Ermittlung des Materialverhaltens ist die Anwendung der Molekulardynamiksimulation.
Fragestellung
Bei der Herstellung und Verarbeitung verschiedenster Materialien bestehen Umgebungsbedingungen, die die Verarbeitungsprozesse beeinflussen. Sowohl bei der Herstellung von Materialien als auch in der Weiterverarbeitung von Halbzeugen entstehen Grenzflächen in der Mikroebene zwischen Atomen und Molekülen. Die makroskopischen Materialeigenschaften werden dabei durch die atomaren Prozesse an der Grenzfläche bestimmt. Besonders bei Materialverbunden ist dieser Aspekt bezogen auf den mechanischen Zusammenhalt von Bedeutung.
Bislang erfolgt die konstruktive Auslegung eines Bauteils hinsichtlich der Materialbeanspruchung weitestgehend auf der makroskopischen Ebene. Die im vorgesehenen Bauteileinsatz auftretenden Belastungen werden experimentell ermittelt und durch Belastungssimulation in der Makroebene überprüft und somit in der Bauteilauslegung berücksichtigt. Die entstehenden Grenzflächeninteraktionen in der Mikroebene werden durch wissenschaftliche Betrachtungen in der Grundlagenforschung beschrieben, bislang aber nur selten in der Materialherstellung oder der konstruktiven Bauteilauslegung berücksichtigt.
Lösung und Umsetzung
Die Betrachtung der Interaktion an der Grenzfläche auf atomarer und molekularer Ebene führt zu einem besseren Verständnis des Einflusses von Umgebungsbedingungen. Die Einflüsse sind physikalische und chemische Vorgänge in der Makroebene. Durch die Anwendung der mikroskopischen Modellierung mittels Molekulardynamik ist die Reduktion der Betrachtungsweise auf die Wechselwirkung zwischen Atomen und Molekülen möglich. Bei der Molekulardynamiksimulation werden Wechselwirkungen zwischen Atomen und Molekülen betrachtet und die sich daraus ergebenden räumlichen Bewegungen ermittelt. Die auftretenden Kräfte werden aus interatomaren Wechselwirkungspotenzialen berechnet. Dabei ist besonders die Betrachtung des Kontakts von Oberflächen von Bedeutung.
Beispielsweise lässt sich mit der molekulardynamischen Simulation der Einfluss der des Sauerstoffs aus der Umgebungsluft auf das Fügen von metallischen Halbzeugen ermitteln. Die Bildung einer Oxidschicht an der Halbzeugoberfläche verändert das physikalisch-chemische Verhalten der Fügepartner. Die Güte der Fügeverbindung wird dadurch maßgeblich beeinflusst. Die Ergebnisse der Molekulardynamiksimulation ermöglichen das Verständnis der Auswirkungen des Sauerstoffeinflusses auf das Fügeergebnis und somit die optimierte Auslegung von Verarbeitungsparametern.
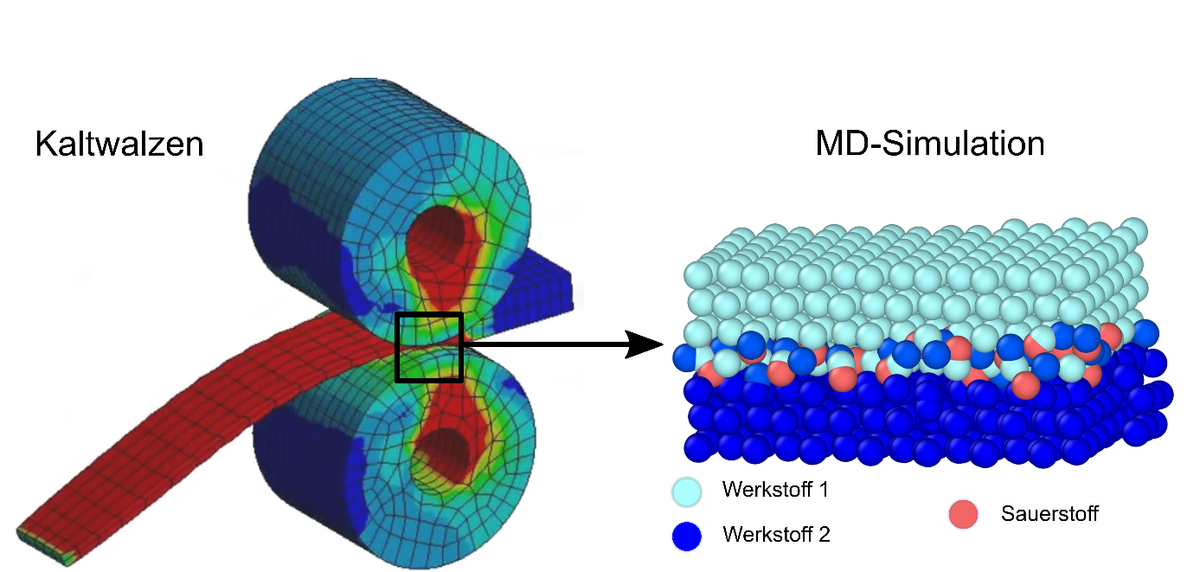
Abb. 1: Schema des Kontaktes zweier Verbundwerkstoffe beim Fügen durch Kaltwalzplattieren (© TU Clausthal)
Für die Durchführung der Simulationen wird die Open-Source-Software LAMMPS (Large-scale Atomic/Molecular Massively-Parallel-Simulator) verwendet. Diese Software ermöglicht die anwendungsspezifische Konfiguration von Simulationsparametern und lässt sich dadurch für viele Problemstellungen anwenden.
Vorteile
Durch die Molekulardynamiksimulation können die Auswirkungen von Umgebungsbedingungen auf das spätere Materialverhalten besser abgeschätzt werden. Eine Erweiterung der bislang angewendeten Material- und Bauteilsimulation ist somit gegeben. Die Simulation wird durch experimentelle und analytische Verfahren begleitet und erreicht somit ein hohes Maß an Praxisrelevanz.
Weitere Anwendungsmöglichkeiten
- Bestimmung der Adhäsionseigenschaften in Abhängigkeit des Sauerstoffanteils bei Fügeverbindungen metallischer Leichtbauwerkstoffe
- Einfluss der Oxidation auf mechanische Eigenschaften von metallischen Schäumen
- Bildung kritischer Verbindungen in Recyclingschlacken beim pyrometallurgischen Recycling von Li-Ionen-Batterien
- Nanoindentation beschichteter Aluminiumoberflächen
- Dynamische Belastungsanalyse bei Hochentropie-Legierungen
- Analyse von Verarbeitungsprozessen metallischer Pulver, wie Additive Fertigung, Beschichten und Fügen
Entwicklungsstand/Erprobungsgrad
Das Anwendungspotenzial der Molekulardynamiksimulation wurde durch die wissenschaftliche Betrachtung verschiedener interdisziplinärer Problemstellungen erfolgreich angewendet.
Schlagworte
Materialeigenschaften, Simulation, Grenzflächen, Berechnung, Metalle, Oxidation, Molekulardynamiksimulation
Weiterführende Informationen
Sonderforschungsbereich 1368: Sauerstofffreie Produktion, Teilprojekt C05 Fügesimulation
Forschungseinrichtung
Prof. Dr. Nina Merkert
TU Clausthal
Simulationswissenschaftliches Zentrum Clausthal-Göttingen


